FASTQC使用
, 18 Dec 2019
fastqc:一种用于高通量序列数据的质量控制应用程序。
安装
- 下载源代码网址
需要
bash #下载后解压 unzip fastqc_*.zip cd fastqc_*.zip chmod 744 fastqc || chmod u+x fastqc# 将 fastqc 设置为可执行程序 #chmod中数字4为设置可读,2可写,1可执行,即r,w,x,而数字7为4+2+1 #u 表示该文件的拥有者,g 表示与该文件的拥有者属于同一个群体(group)者,o 表示其他以外的人,a 表示这三者皆是 - conda 安装就好了,自动帮下jdk.
–help
fastqc –help
# 命令行使用
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] [-c contaminant file] seqfile1 .. seqfileN
参数说明
-h --help
-v --version
-o -output dir
- casave 文件来自原始 casave 输出
-nano 文件来自 naopore 序列,采用 fast5 格式
-extract 如果设置,则压缩输出
-j --java java二进制文件完整路径
-nogroup 禁止读取2500bp以上的碱基组
-f 跳过正常文件格式检测,强制使用指定格式 bam | sam | bam_mapped | sam_mapped | fastq
-t --threads 多线程,每个线程 250 M
-c --contamin 指定包含列表的非默认文件,污染物筛选过多的序列(哈希)
-a -adapters 指定包含列表的非默认文件,包含一组已经命名的Adapter(哈希)
-l 指定一个非默认文件,限制将用于确认 warning / Fairure,或者从结果中删除一些模块, cofiguration --> limits.txt
-k -kmers 指定要在Kmer中查找的长度,必须在2-10 之间,默认为7
-q -quiet 安静模式,在标准输出上禁止所有的进度消息,只报错
-d --dir 一个目录用于写入临时文件当生成图像时, 默认系统临时目录
生成文件解读
当使用1个fastq文件时,使用命令mkdir fastqc && fastqc -o ./fastqc ref.fastq.gz
会在目录下生成一个html以及.zip文件,zip文件就是画图的数值以及图片。所以fastqc结果看html即可。
- html解读
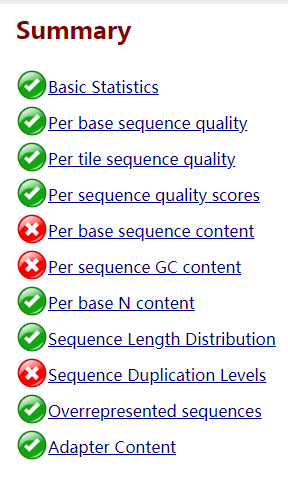 这一部分中√代表”PASS”;!代表”WARN”;x代表”FAIL”
1.Basic Statics
这一部分中√代表”PASS”;!代表”WARN”;x代表”FAIL”
1.Basic Statics
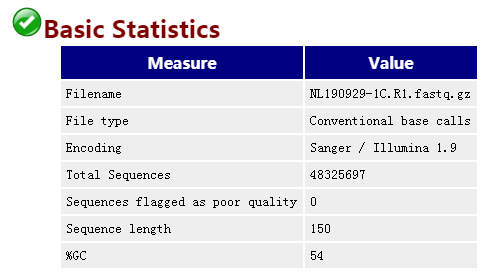
Encoding为Illumina1.9 就是 Phred+33,很重要,1.8以上即为Phred33编码。
Total sequences: reads数量(reads就是高通量测序平台产生的序列标签,翻译为读段)
Sequence length: 测序长度
%GC: GC含量: 重点关注,可以帮助区别物种,人类细胞42%左右 2.Per base sequence quality
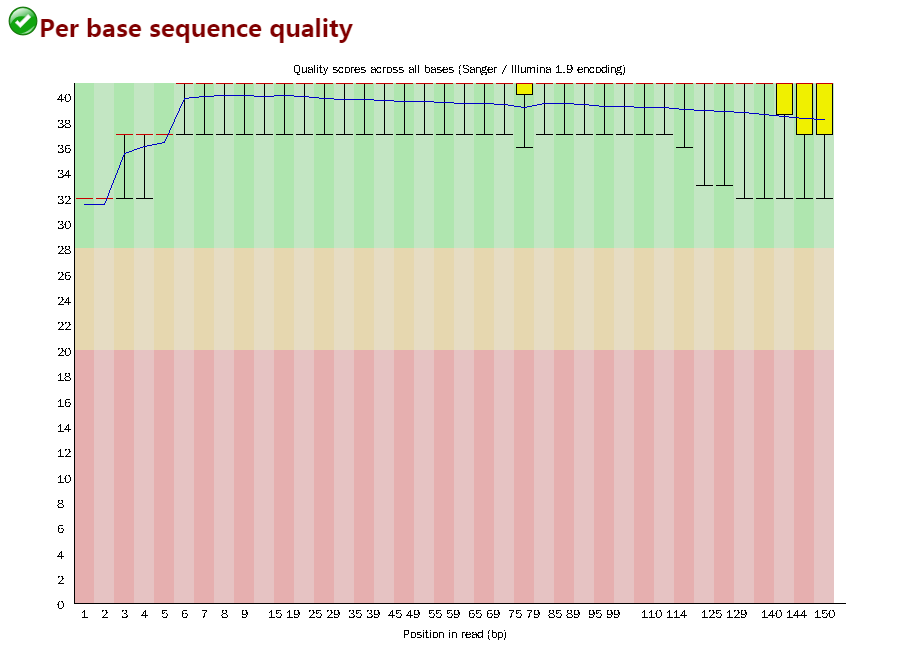 画的是boxplot,
画的是boxplot,这个图我看了下数值,前面的绘图数据都是32。 横轴:测序序列的1-150个碱基; 纵轴:质量得分,score = -10 * log10(error),例如错误率error为1%,那么算出的score就是20 蓝色的线将各个碱基的质量平均值连接起来